Все мы знаем кто надувает воздушные шарики и делает людям при его вдыхании такой смешной голос. Конечно же это гелий. 2-й в таблице Менделеева.
Гелий-один из наиб. распространенных элементов космоса-занимает второе место после водорода. Содержание гелия в атмосфере (образуется в результате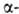 распада Ac, Th, U) 5,27*10-4% по объему. Запасы гелия в атмосфере, литосфере и гидросфере оцениваются в 5*1014 м3. Гелионосные прир. газы содержат, как правило, до 2% по объему гелия; главные пром. месторождения этих газов находятся в США (2,1*1010 м3 Т.), СССР, Канаде (108 м3), ЮАР. Гелий содержится также в минералах: клевеите, монаците, торианите (до 10,5 л/кг).
распада Ac, Th, U) 5,27*10-4% по объему. Запасы гелия в атмосфере, литосфере и гидросфере оцениваются в 5*1014 м3. Гелионосные прир. газы содержат, как правило, до 2% по объему гелия; главные пром. месторождения этих газов находятся в США (2,1*1010 м3 Т.), СССР, Канаде (108 м3), ЮАР. Гелий содержится также в минералах: клевеите, монаците, торианите (до 10,5 л/кг).
распада Ac, Th, U) 5,27*10-4% по объему. Запасы гелия в атмосфере, литосфере и гидросфере оцениваются в 5*1014 м3. Гелионосные прир. газы содержат, как правило, до 2% по объему гелия; главные пром. месторождения этих газов находятся в США (2,1*1010 м3 Т.), СССР, Канаде (108 м3), ЮАР. Гелий содержится также в минералах: клевеите, монаците, торианите (до 10,5 л/кг).Свойства. Гелий-одноатомный газ без цвета и запаха. Св-ва изотопов гелия приведены в таблице. Ур-ния температурной зависимости давления пара 4Не:

Теплопроводность гелия 0,1437 Вт/(м*К); магн. восприимчивость — 1,9*10-6; ур-ние температурной зависимости вязкости: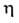 *107 = 5,023Т0,647 Па*с (4-1000 К).
*107 = 5,023Т0,647 Па*с (4-1000 К).
*107 = 5,023Т0,647 Па*с (4-1000 К).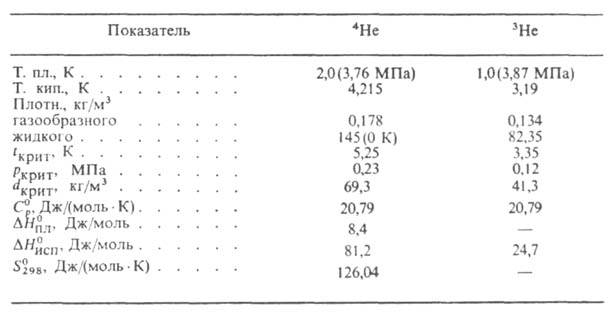
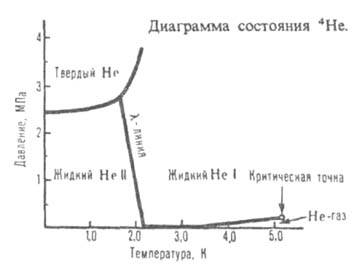
Жидкий гелий-квантовая жидкость, т.е. жидкость, в макроскопич. объеме к-рой проявляются квантовые св-ва составляющих ее атомов. Квантовые эффекты существенны при очень низких т-рах, когда длина волны де Бройля для теплового движения атомов становится сравнимой с расстоянием между ними. На рисунке приведена диаграмма состояния 4Не. При 2,17 К и давлении паров 0,005 МПа (т. наз.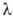 -точка) жидкий 4Не (бозе-жидкость) претерпевает фазовый переход второго рода (от Не I к Не II), сопровождающийся резким изменением ряда св-в: теплоемкости, вязкости, плотности и др. С увеличением давления т-ра перехода
-точка) жидкий 4Не (бозе-жидкость) претерпевает фазовый переход второго рода (от Не I к Не II), сопровождающийся резким изменением ряда св-в: теплоемкости, вязкости, плотности и др. С увеличением давления т-ра перехода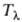 смещается в область более низких т-р (зависимость
смещается в область более низких т-р (зависимость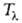 от давления показана на рисунке
от давления показана на рисунке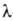 -линией). Для Не II характерна сверхтекучесть-способность протекать без трения через узкие (диам. менее 100 нм) капилляры и щели. Это св-во открыто в 1938 П.Л. Капицей. Сверхтекучесть обусловлена переходом при т-рах ниже
-линией). Для Не II характерна сверхтекучесть-способность протекать без трения через узкие (диам. менее 100 нм) капилляры и щели. Это св-во открыто в 1938 П.Л. Капицей. Сверхтекучесть обусловлена переходом при т-рах ниже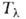 части атомов жидкого гелия в состояние с нулевым импульсом. Не I бурно кипит во всем объеме, Не II-спокойная жидкость с ясно выраженным мениском. Различие в их поведении объясняется необычайно высокой теплопроводностью Не II (во много миллионов раз выше, чем у Не I). Сверхтекучесть проявляет также и жидкий 3Не (ферми-жидкость) вблизи абс. нуля (менее 2,6*10-3 К) и давлении ок. 3,4 МПа. Жидкий гелий-единственное в-во, не затвердевающее при нормальном давлении даже вблизи О К. Он кристаллизуется только под давлением более 2,5 МПа. Кристаллич. решетка 4Не гексагональная с плотной упаковкой. 3Не при одной и той же т-ре в зависимости от давления может находиться в двух модификациях:
части атомов жидкого гелия в состояние с нулевым импульсом. Не I бурно кипит во всем объеме, Не II-спокойная жидкость с ясно выраженным мениском. Различие в их поведении объясняется необычайно высокой теплопроводностью Не II (во много миллионов раз выше, чем у Не I). Сверхтекучесть проявляет также и жидкий 3Не (ферми-жидкость) вблизи абс. нуля (менее 2,6*10-3 К) и давлении ок. 3,4 МПа. Жидкий гелий-единственное в-во, не затвердевающее при нормальном давлении даже вблизи О К. Он кристаллизуется только под давлением более 2,5 МПа. Кристаллич. решетка 4Не гексагональная с плотной упаковкой. 3Не при одной и той же т-ре в зависимости от давления может находиться в двух модификациях: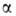 (решетка кубическая) и
(решетка кубическая) и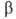 (гексагональная с плотной упаковкой); т-ра тройной точки
(гексагональная с плотной упаковкой); т-ра тройной точки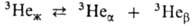 3,15 К, давл. 14,3 МПа; у 4Не тройная точка отсутствует.
3,15 К, давл. 14,3 МПа; у 4Не тройная точка отсутствует.
-точка) жидкий 4Не (бозе-жидкость) претерпевает фазовый переход второго рода (от Не I к Не II), сопровождающийся резким изменением ряда св-в: теплоемкости, вязкости, плотности и др. С увеличением давления т-ра перехода смещается в область более низких т-р (зависимостьот давления показана на рисунке-линией). Для Не II характерна сверхтекучесть-способность протекать без трения через узкие (диам. менее 100 нм) капилляры и щели. Это св-во открыто в 1938 П.Л. Капицей. Сверхтекучесть обусловлена переходом при т-рах нижечасти атомов жидкого гелия в состояние с нулевым импульсом. Не I бурно кипит во всем объеме, Не II-спокойная жидкость с ясно выраженным мениском. Различие в их поведении объясняется необычайно высокой теплопроводностью Не II (во много миллионов раз выше, чем у Не I). Сверхтекучесть проявляет также и жидкий 3Не (ферми-жидкость) вблизи абс. нуля (менее 2,6*10-3 К) и давлении ок. 3,4 МПа. Жидкий гелий-единственное в-во, не затвердевающее при нормальном давлении даже вблизи О К. Он кристаллизуется только под давлением более 2,5 МПа. Кристаллич. решетка 4Не гексагональная с плотной упаковкой. 3Не при одной и той же т-ре в зависимости от давления может находиться в двух модификациях: (решетка кубическая) и (гексагональная с плотной упаковкой); т-ра тройной точки 3,15 К, давл. 14,3 МПа; у 4Не тройная точка отсутствует.Для газообразного гелия характерна высокая способность проникать сквозь перегородки из пластмасс, стекла и нек-рых металлов. Р-римость гелия: в воде(смэ/л)-9,78 (0°С), 8,61 (20°С), 10,10 (80°С); этаноле (% по объему)-2,8 (15°С), 3,2 (25 °С). Гелий характеризуется исключительной хим. инертностью.
Получение. Гелий выделяют из прир. гелионосных горючих газов. Сухой газ, очищенный от СО2, под давл. 2 МПа подается в систему теплообменников и сепараторов, где благодаря конденсации при —28, —41 и — 110°С отделяется значит. часть углеводородов. Полученная парожидкостная смесь дросселируется до давл. 1,2 МПа и в результате отделения жидкой фазы парогазовая смесь обогащается гелием до содержания 3%. При послед.дросселировании до 1,0 МПа происходит дальнейшее обогащение-сначала до содержания 30-50% гелия, затем при охлаждении кипящим при - 203°С и 0,04 МПа азотом-до 90%. Сырой гелий (70-90% по объему гелия) очищают от водорода (4-5%) с помощью СиО при 650-800 К, а затем осушают в адсорберахсиликагелем. Окончательная очистка достигается охлаждением сырого гелия кипящим под вакуумом N2 и адсорбцией примесей на активном угле в адсорберах, также охлаждаемых жидким N2. Производят гелий техн. чистоты (99,80% по объему гелия) и высокой чистоты (99,985%).
Определение. Качественно гелий обнаруживают с помощью эмиссионного спектрального анализа; осн. характеристич. линии 587,56 и 388,86 нм. Для количеств. определения пользуются методами, основанными на измерении физ. св-в (плотности, теплопроводности и др.), а также масс-спектро-метрией игазовой хроматографией.
Применение. Газообразный гелий применяют: в кач-ве защитной среды при сварке, резке и плавке металлов (15% производимого гелия), при произ-ве твэлов (25%), при перекачивании ракетного топлива (35%), в произ-ве полупроводниковых материалов; для консервации пищ. продуктов; в кач-ве теплоносителя в высокотемпературных ядерных реакторах; для заполнения дирижаблей и аэростатов; в вакуумной технике в кач-ве рабочей среды при обнаружении течей гелиевым течеискателем; как компонент среды газовых лазеров; газ-носитель в хроматографии; термометрич. в-во в газовых эталонных термометрах в интервале т-р 1-80 К; компонент дыхат. смесей для глубоководного погружения. Жидкий гелий-хладагент в эксперим. физике, при получении сверхпроводящих материалов для вычислит. и измерит, техники, в сверхпроводящих магнитах и др. Жидкий 3Не-термометрич. в-во для измерений т-р ниже 1 К.
Мировое произ-во гелия ок. 54 млн. м3 (1979). Осн. страны-производители-США, СССР, ЮАР.
Хранится гелий под давл. 15 МПа в стальных баллонах емкостью 40 л, окрашенных в коричневый цвет; жидкий гелий-в дьюаровских сосудах емкрстью 10 л с защитным экраном, охлаждаемым жидким N2. Гелий открыт в 1868 Ж. Жансеном и Н. Локьером в спектре солнечной короны; впервые выделен в 1895 У. Рамзаем из минерала клевеита.
Словарик:
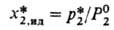
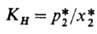
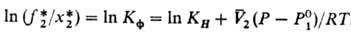
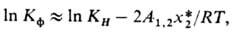
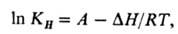
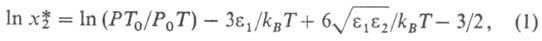
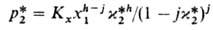
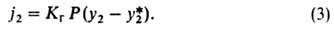
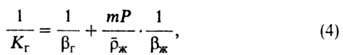
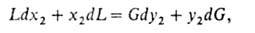
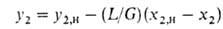
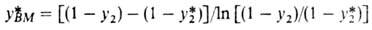
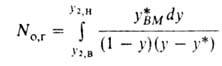
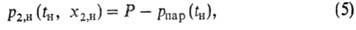
Словарик:
АБСОРБЦИЯ газов (лат. absorptio, от absorbeo-поглощаю), объемное поглощение газов и паров жидкостью (абсорбентом) с образованием р-ра. Применение абсорбции в технике для разделения и очистки газов, выделения паров из паро-газовых смесей основано на разл. р-римости газов и паров в жидкостях. Процесс, обратный абсорбции., наз. десорбцией; его используют для выделения из р-ра поглощенного газа и регенерации абсорбента. Поглощение газовметаллами (напр., водорода палладием) наз. окклюзией. Абсорбция - частный случай сорбции.
Различают физ. и хим. абсорбцию. При физической абсорбции энергия взаимод. молекул газа и абсорбента в р-ре не превышает 20 кДж/моль. При химической абсорбции (или абсорбция с хим. р-цией, часто наз. хемосорбцией) молекулы растворенного газа реагируют с активным компонентом абсорбента-хемосорбентом (энергия взаимод. молекул более 25 кДж/моль) либо в р-ре происходит диссоциация или ассоциация молекул газа. Промежут. варианты абсорбции характеризуются энергией взаимод. молекул 20-30 кДж/моль. К таким процессам относится растворение с образованием водородной связи, в частности абсорбция ацетилена диметилформамидом.
Статика абсорбции. Характеризует термодинамич. равновесие р-ра с паро-газовой смесью, а также материальный и энергетич. балансы процесса. При физ. абсорбции с образованием идеального р-ра для р-рите-ля и растворенного газа во всем интервале изменения состава в соответствии с законом Раулярастворимость газа:
где Р°2- давление паров над чистым сжиженным газом при данной т-ре системы; р2 - парциальное давление газа; звездочкой обозначаются параметры в-ва в условиях равновесия. Индексы "1" и "2" относятся соотв. к р-рителю и газу. Идеальная р-римость x*2,ид-ф-ция только т-ры, св-ва р-ри-теля влияния на нее не оказывают.
Зависимость р-римости газов х*2 от их парциального давления при физ. абсорбции в бесконечно разбавленном р-ре (х2 ->0) и при низких давлениях Р в системе описывается законом Генри (рис. 1, прямые 1-3):
где КH-коэф. Генри, изменяющийся с изменением т-ры. Если абсорбция проводят под давлением, но х*2 ->0, р-римость газа можно рассчитать по уравнению Кричевского - Казарновского:
где Кф-коэф. физ. р-римости, равный КH при х2-> 0 и Р-> -> 0; f*2-летучесть газа;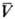 -парциальный мольный объем растворенного газа в жидкой фазе при бесконечном разбавлении; R- универсальная газовая постоянная; Po1-давление насыщ. паров чистого р-рителя при абс. т-ре системы Т. Если 0 < х*2
-парциальный мольный объем растворенного газа в жидкой фазе при бесконечном разбавлении; R- универсальная газовая постоянная; Po1-давление насыщ. паров чистого р-рителя при абс. т-ре системы Т. Если 0 < х*2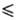 (0,05-0,1) молярной доли (разбавленные р-ры), то при низких давлениях справедливо ур-ние Сеченова:
(0,05-0,1) молярной доли (разбавленные р-ры), то при низких давлениях справедливо ур-ние Сеченова:
-парциальный мольный объем растворенного газа в жидкой фазе при бесконечном разбавлении; R- универсальная газовая постоянная; Po1-давление насыщ. паров чистого р-рителя при абс. т-ре системы Т. Если 0 < х*2(0,05-0,1) молярной доли (разбавленные р-ры), то при низких давлениях справедливо ур-ние Сеченова:где А1,2-коэф., не зависящий от состава р-ра.
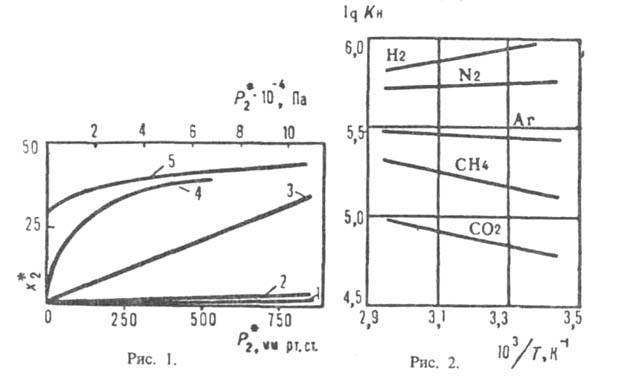
Рис. 1. Зависимость р-римости х2* нек-рых газов в жидкостях парциального давления газов над растворами: I- СО2 в воде при 20 °С; 2-СО2 в пропиленкар-бонате при 25 С; 3 С2Н4 в диметилформамиде при 25 °С; 4-СО, в водном о-ое содержащем 25% К2СО3 и 10% диэтаноламина при 60°C 5-СО, в 2,5 н водном р-ре моноэтаноламина при 20°С; х2* - в м3 газа (при нормальных условиях - 20°С и 0,1 МПа) на 1 м3 жидкости. 2
Зависимость растворимости газа от температуры как при физ., так и при хим. абсорбции приближенно описывается ур-нием:
где -тепловой эффект растворения газа. Если р-р при абсорбции нагревается,
-тепловой эффект растворения газа. Если р-р при абсорбции нагревается,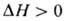 . С изменением т-ры
. С изменением т-ры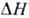 обычно либо остается постоянной, либо незначительно изменяется. Р-римость газа в смешанном растворителе (содержащем малополярные компоненты) можно оценить по соотношению:
обычно либо остается постоянной, либо незначительно изменяется. Р-римость газа в смешанном растворителе (содержащем малополярные компоненты) можно оценить по соотношению:
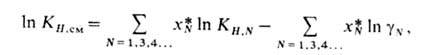
-тепловой эффект растворения газа. Если р-р при абсорбции нагревается, . С изменением т-ры обычно либо остается постоянной, либо незначительно изменяется. Р-римость газа в смешанном растворителе (содержащем малополярные компоненты) можно оценить по соотношению:где КН.си, КН.N-коэф. Генри соотв. для газа в смеси р-рителей и для р-ров этого газа в чистых компонентах р-рителя;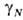 -коэф. активности компонентов р-рителя (молярные доли
-коэф. активности компонентов р-рителя (молярные доли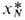 относятся к смеси р-рителей, свободной от растворенного газа).
относятся к смеси р-рителей, свободной от растворенного газа).
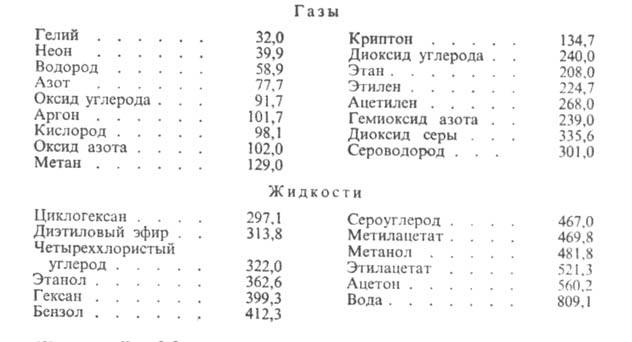
-коэф. активности компонентов р-рителя (молярные долиотносятся к смеси р-рителей, свободной от растворенного газа).Нек-рые данные о р-римости газов приведены в таблице, где газы и р-рители расположены в порядке возрастания энергетич. параметров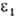 и
и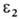 потенциала Леннард-Джонса. Эти параметры м. б. использованы для приближенной оценки р-римости газов при низких давлениях по ур-нию:
потенциала Леннард-Джонса. Эти параметры м. б. использованы для приближенной оценки р-римости газов при низких давлениях по ур-нию:
и потенциала Леннард-Джонса. Эти параметры м. б. использованы для приближенной оценки р-римости газов при низких давлениях по ур-нию:где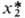 -р-римость в 1 м3 газа, приведенная к нормальным условиям (20 °С, 0,1 МПа) на 1 м3 абсорбента; kв- константа Больцмана; Р0, T0-соотв. давление и т-ра при нормальных условиях; Р, Г-то же при рабочих условиях. Р-римость умеренно растворимых газов в данном р-рителе возрастает линейно с увеличением
-р-римость в 1 м3 газа, приведенная к нормальным условиям (20 °С, 0,1 МПа) на 1 м3 абсорбента; kв- константа Больцмана; Р0, T0-соотв. давление и т-ра при нормальных условиях; Р, Г-то же при рабочих условиях. Р-римость умеренно растворимых газов в данном р-рителе возрастает линейно с увеличением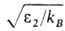
-р-римость в 1 м3 газа, приведенная к нормальным условиям (20 °С, 0,1 МПа) на 1 м3 абсорбента; kв- константа Больцмана; Р0, T0-соотв. давление и т-ра при нормальных условиях; Р, Г-то же при рабочих условиях. Р-римость умеренно растворимых газов в данном р-рителе возрастает линейно с увеличением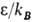 (в К) для
(в К) для 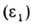 и
и 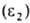 :
:
Тепловой эффект растворения газа АЯ линейно изменяется с увеличением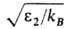 ; соотв. р-римость плохо р-римых газов
; соотв. р-римость плохо р-римых газов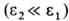 , в основном Не, Ne, H2, N2, CO, Аr, О2 и NO, с возрастанием т-ры увеличивается (за исключением водных р-ров), а р-римость хорошо р-римых газов
, в основном Не, Ne, H2, N2, CO, Аr, О2 и NO, с возрастанием т-ры увеличивается (за исключением водных р-ров), а р-римость хорошо р-римых газов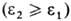 уменьшается. Типичные примеры для бесконечно разбавленных р-ров приведены на рис. 2. Р-римость таких газов, как H2S, COS, SO2, HC1, NH3, C12, обычно значительно выше, чем рассчитанная по ур-нию (1), вследствие специфич. взаимод. с молекулами р-рителя.
уменьшается. Типичные примеры для бесконечно разбавленных р-ров приведены на рис. 2. Р-римость таких газов, как H2S, COS, SO2, HC1, NH3, C12, обычно значительно выше, чем рассчитанная по ур-нию (1), вследствие специфич. взаимод. с молекулами р-рителя.
; соотв. р-римость плохо р-римых газов, в основном Не, Ne, H2, N2, CO, Аr, О2 и NO, с возрастанием т-ры увеличивается (за исключением водных р-ров), а р-римость хорошо р-римых газов уменьшается. Типичные примеры для бесконечно разбавленных р-ров приведены на рис. 2. Р-римость таких газов, как H2S, COS, SO2, HC1, NH3, C12, обычно значительно выше, чем рассчитанная по ур-нию (1), вследствие специфич. взаимод. с молекулами р-рителя.При хим. абсорбции поглотит. способность абсорбента (емкость, соответствующая предельному кол-ву газа, к-рый поглощается единицей объема абсорбента) и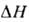 больше, чем при физ. абсорбции. При необратимой р-ции (напр., при поглощении СО2 р-рами NaOH с образованием Na2CO3) равновесное давление газанад р-ром равно нулю, пока в р-ре есть непрореагировавший абсорбент, и поглотит. способность определяется стехиометрией р-ции. При обратимой р-циидавление газа над р-ром равно нулю, но по сравнению с физ. абсорбцией резко изменяется характер зависимости р-римости газа от давления (рис. 1, кривые 4,5). Так в простейшем случае, когда в р-ре происходит только одна р-ция и активности компонентов р-ра равны их концентрациям, имеем:
больше, чем при физ. абсорбции. При необратимой р-ции (напр., при поглощении СО2 р-рами NaOH с образованием Na2CO3) равновесное давление газанад р-ром равно нулю, пока в р-ре есть непрореагировавший абсорбент, и поглотит. способность определяется стехиометрией р-ции. При обратимой р-циидавление газа над р-ром равно нулю, но по сравнению с физ. абсорбцией резко изменяется характер зависимости р-римости газа от давления (рис. 1, кривые 4,5). Так в простейшем случае, когда в р-ре происходит только одна р-ция и активности компонентов р-ра равны их концентрациям, имеем:
больше, чем при физ. абсорбции. При необратимой р-ции (напр., при поглощении СО2 р-рами NaOH с образованием Na2CO3) равновесное давление газанад р-ром равно нулю, пока в р-ре есть непрореагировавший абсорбент, и поглотит. способность определяется стехиометрией р-ции. При обратимой р-циидавление газа над р-ром равно нулю, но по сравнению с физ. абсорбцией резко изменяется характер зависимости р-римости газа от давления (рис. 1, кривые 4,5). Так в простейшем случае, когда в р-ре происходит только одна р-ция и активности компонентов р-ра равны их концентрациям, имеем:где Кх = ASKфKP-константа равновесия системы газ-жидкость; Кр — константа равновесия р-ции;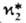
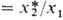 -равновесная степень превращения абсорбента; x1- начальная концентрация абсорбента; h-число молей продуктов р-ции на 1 моль прореагировавшего газа; j-число молей абсорбента, вступивших во взаимод. с 1 молем растворенного газа; A5-коэф., зависящий от стехиометрии.
-равновесная степень превращения абсорбента; x1- начальная концентрация абсорбента; h-число молей продуктов р-ции на 1 моль прореагировавшего газа; j-число молей абсорбента, вступивших во взаимод. с 1 молем растворенного газа; A5-коэф., зависящий от стехиометрии.
-равновесная степень превращения абсорбента; x1- начальная концентрация абсорбента; h-число молей продуктов р-ции на 1 моль прореагировавшего газа; j-число молей абсорбента, вступивших во взаимод. с 1 молем растворенного газа; A5-коэф., зависящий от стехиометрии.Коэф. ускорения абсорбции могут быть достаточно велики. Так, в случае поглощения СО2 в насадочной колонне при одинаковых нагрузках по фазам, т-ре идавлении, используя 2 н. водный р-р КОН (15% К содержится в р-ре в виде карбоната), можно получить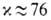 по сравнению с физ. абсорбцией СО2 водой. Гипотетич. идеальный р-ритель, не обладающий сопротивлением переносу в жидкой фазе и имеющий бесконечно большую реакц. способность, обеспечил бы
по сравнению с физ. абсорбцией СО2 водой. Гипотетич. идеальный р-ритель, не обладающий сопротивлением переносу в жидкой фазе и имеющий бесконечно большую реакц. способность, обеспечил бы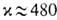
по сравнению с физ. абсорбцией СО2 водой. Гипотетич. идеальный р-ритель, не обладающий сопротивлением переносу в жидкой фазе и имеющий бесконечно большую реакц. способность, обеспечил быУвеличение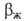 и
и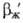 (иногда в неск. раз) может происходить под влиянием поверхностной конвекции, вызываемой локальными градиентами поверхностного натяжения, к-рые возникают в ряде случаев в результате массоотдачи, особенно при одноврем. протекании р-ций (напр., при абсорбции СО2 водными р-рамимоноэтаноламина). Это необходимо учитывать при подборе новых хемосорбентов. Значение
(иногда в неск. раз) может происходить под влиянием поверхностной конвекции, вызываемой локальными градиентами поверхностного натяжения, к-рые возникают в ряде случаев в результате массоотдачи, особенно при одноврем. протекании р-ций (напр., при абсорбции СО2 водными р-рамимоноэтаноламина). Это необходимо учитывать при подборе новых хемосорбентов. Значение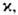 если р-ция приводит к возникновению поверхностной конвекции, следует определять на основе коэф. массоотдачи при физ. абсорбции, найденного в условиях воздействия на процесс конвективных микропотоков вблизи границы раздела фаз.
если р-ция приводит к возникновению поверхностной конвекции, следует определять на основе коэф. массоотдачи при физ. абсорбции, найденного в условиях воздействия на процесс конвективных микропотоков вблизи границы раздела фаз.
и (иногда в неск. раз) может происходить под влиянием поверхностной конвекции, вызываемой локальными градиентами поверхностного натяжения, к-рые возникают в ряде случаев в результате массоотдачи, особенно при одноврем. протекании р-ций (напр., при абсорбции СО2 водными р-рамимоноэтаноламина). Это необходимо учитывать при подборе новых хемосорбентов. Значение если р-ция приводит к возникновению поверхностной конвекции, следует определять на основе коэф. массоотдачи при физ. абсорбции, найденного в условиях воздействия на процесс конвективных микропотоков вблизи границы раздела фаз.При расчете скорости абсорбции часто используют коэф. массо-передачи, определяемые по гипотетич. поверхностным составам и, следовательно, по гипотетич. движущим силам. Обычно принимают, что коэф. массопередачи, отнесенный к концентрации в газе, Кг [кмоль/(м2 *МПа*с)] обусловлен движущей силой (у2-у*2), где у*2-молярная доля поглощаемого компонента в газе, к-рая отвечает равновесию с жидкостью, имеющей средний объемный состав х2; у2 -средний объемный состав газа в данном сечении аппарата. Тогда получим:
Аналогично можно найти движущую силу (x*2 — х2) и коэф. массопередачи Кж. Из выражений (2) и (3) следует:
где т = (y2,гр — y*2)/(x2,гр - х2)-наклон равновесной линии в интервале концентраций от х2, у2до x2,гр, y2,гр. Выражение (4) записано для локального коэф. массопередачи и показывает, что этот коэффициент зависит от наклона линии равновесия. Наиб. удобно рассчитывать коэф. массопередачи по ур-нию (4) в случаях, когда наклон равновесной линии остается почти постоянным в рабочем интервале концентраций. При искривленной линии равновесия необходимо учитывать зависимость m от концентрации.
Абсорбцию осуществляют в массообменных аппаратах, наз. абсорберами,-тарельчатых, насадочных (устаревшее название-скрубберы), пленочных, роторно-пленочных и распылительных. Схема материальных потоков в абсорбере представлена на рис. 3. Связь между концентрациями поглощаемого компонента вгазе у2 и в жидкости в любом горизонтальном сечении аппарата находят из ур-ния материального баланса (т. наз. ур-ние рабочей линии). В общем случае это ур-ние имеет вид:
где L и G-расходы жидкости и газа. Когда объемы фаз в ходе абсорбции изменяются незначительно, рабочая линия-прямая:
Здесь индексом "н" обозначается ниж. сечение противоточного абсорбера или десорбера.
Существенное влияние на ход рабочей и равновесной [у* =f(x*2)] линий могут оказать тепловые эффекты абсорбции. Ход рабочей линии может сильно зависеть от интенсивности испарения р-рителя (особенно при десорбции). Если абсорбция сопровождается значит. выделением теплоты, а кол-во абсорбированного в-ва достаточно велико, р-ритель может сильно нагреваться при прохождении через колонну. Примеры-осушка воздуха с помощью конц. H2SO4, растворение НС1 в воде при получении конц. соляной к-ты. Температурный режим абсорбера, от к-рого зависят равновесное давление поглощаемого компонента, т.е. движущая сила процесса, физ.-хим. св-ва системы и ход рабочей линии рассчитывают по ур-нию теплового баланса абсорбера.
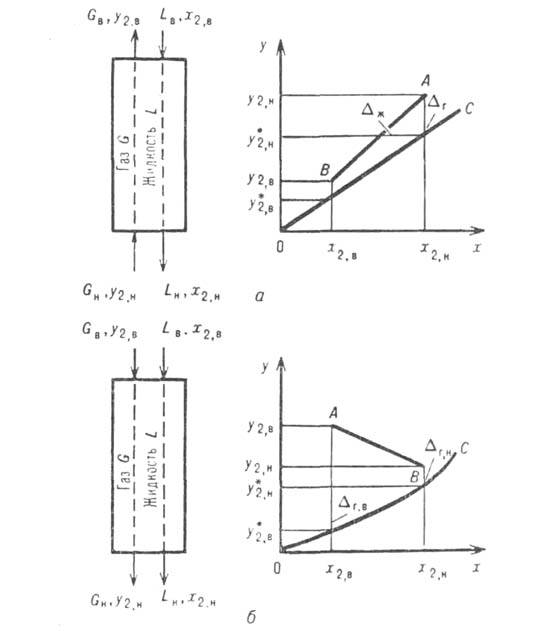
Рис. 3. Схема материальных потоков в абсорбере и хол рабочей и равновесной линий (а-при противотоке, 6-при прямотоке): ЛВ-рабочая линия; ОС-равновесная линия; и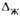 -движущая сила соотв. в газовой фазе в верх, и ниж. сечениях
-движущая сила соотв. в газовой фазе в верх, и ниж. сечениях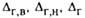 аосороера и в газовой и жидкой фазах на ступени.
аосороера и в газовой и жидкой фазах на ступени.
-движущая сила соотв. в газовой фазе в верх, и ниж. сечениях аосороера и в газовой и жидкой фазах на ступени.При отсутствии внеш. подвода или отвода теплоты, при одинаковых т-рах газа и жидкости и без учета испарения и конденсации абсорбента и теплотрастворения др. газов изменение т-ры абсорбента в любом сечении абсорбера составляет: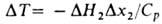 , где Ср - теплоемкость р-ра,
, где Ср - теплоемкость р-ра,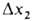 -изменение концентрации газа в рассматриваемом сечении. Обычно принимают, что т-ра жидкости на межфазной границе и в объеме одинаковая. Поскольку наиб. концентрация растворенного газа и соотв. наиб. тепловыделение наблюдаются вблизи пов-сти контакта фаз, т-ра межфазной пов-сти, определяющая истинное равновесие, часто существенно отличается от т-ры объема жидкости. Методы учета этого явления разрабатываются.
-изменение концентрации газа в рассматриваемом сечении. Обычно принимают, что т-ра жидкости на межфазной границе и в объеме одинаковая. Поскольку наиб. концентрация растворенного газа и соотв. наиб. тепловыделение наблюдаются вблизи пов-сти контакта фаз, т-ра межфазной пов-сти, определяющая истинное равновесие, часто существенно отличается от т-ры объема жидкости. Методы учета этого явления разрабатываются.
, где Ср - теплоемкость р-ра, -изменение концентрации газа в рассматриваемом сечении. Обычно принимают, что т-ра жидкости на межфазной границе и в объеме одинаковая. Поскольку наиб. концентрация растворенного газа и соотв. наиб. тепловыделение наблюдаются вблизи пов-сти контакта фаз, т-ра межфазной пов-сти, определяющая истинное равновесие, часто существенно отличается от т-ры объема жидкости. Методы учета этого явления разрабатываются.Чтобы вычислить пов-сть массообмена F, необходимую для обеспечения желаемого изменения состава газа в абсорбере, можно использовать локальные значения скорости массопередачи [см. ур-ние (3)1 совместно с ур-нием материального баланса по абсорбируемому компоненту. При постоянстве коэф. массоперелачи по высоте аппарата:
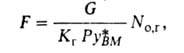
где G-мольная массовая скорость газа, кмоль/(м2*с);
No,r-общее число единиц переноса в газовой фазе:
Этот важный параметр зависит только от технол. режима процесса, определяется положением рабочей и равновесной линий и показывает, как влияет движущая сила абсорбции на высоту аппарата. Число единиц переноса, а следовательно, и высота абсорбера, бесконечно велики, если абсорбер работает при миним. кол-ве циркулирующего абсорбента, когда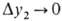 . При увеличении
. При увеличении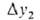 габариты аппарата уменьшаются, но возрастают расход энергии и степеньрастворения плохо растворимых компонентов газовой смеси, что приводит либо к их потере и загрязнению извлекаемого газа, либо к дополнит. затратам на разделение растворенных газов.
габариты аппарата уменьшаются, но возрастают расход энергии и степеньрастворения плохо растворимых компонентов газовой смеси, что приводит либо к их потере и загрязнению извлекаемого газа, либо к дополнит. затратам на разделение растворенных газов.
. При увеличении габариты аппарата уменьшаются, но возрастают расход энергии и степеньрастворения плохо растворимых компонентов газовой смеси, что приводит либо к их потере и загрязнению извлекаемого газа, либо к дополнит. затратам на разделение растворенных газов.При расчете абсорберов, особенно тарельчатых, часто используют понятие эффективности ступени, или степени приближения к равновесию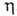 . Эту величину можно определить как отношение фактически реализованного изменения состава к изменению, к-рое произошло бы при достижении равновесия:
. Эту величину можно определить как отношение фактически реализованного изменения состава к изменению, к-рое произошло бы при достижении равновесия: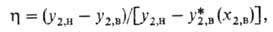
. Эту величину можно определить как отношение фактически реализованного изменения состава к изменению, к-рое произошло бы при достижении равновесия:где индексом "в" обозначается верх. сечение противоточного аппарата.
Во мн. типах ступенчатых контактных устройств достигнута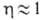 . Это означает, что при мат. анализе таких устройств правомерно использовать понятие о равновесной ступени. Рассчитав число теоретич. тарелок и зная эффективность ступени
. Это означает, что при мат. анализе таких устройств правомерно использовать понятие о равновесной ступени. Рассчитав число теоретич. тарелок и зная эффективность ступени , можно определить число реальных ступеней, необходимых для обеспечения заданной степени разделения.
, можно определить число реальных ступеней, необходимых для обеспечения заданной степени разделения.
. Это означает, что при мат. анализе таких устройств правомерно использовать понятие о равновесной ступени. Рассчитав число теоретич. тарелок и зная эффективность ступени, можно определить число реальных ступеней, необходимых для обеспечения заданной степени разделения.Основы технологии абсорбционных процессов. Абсорбцию часто осуществляют в виде абсорбционно-десорбционного цикла (циклич. процесс), однако стадия десорбции может отсутствовать, если в результате абсорбции получают готовый продукт или регенерация поглотителя невозможна (разомкнутый процесс). На рис. 4 приведена одна из простейших схем абсорбционного разделения газов. Для снижения расхода энергии иногда применяют двух- и многопоточные схемы с отводом грубо- и тонкорегенерированного р-ров в разных сечениях десорбера и подачей их в разл. точки абсорбера либо направляют насыщ. р-р абсорбента в разные точки десорбера и т.п.
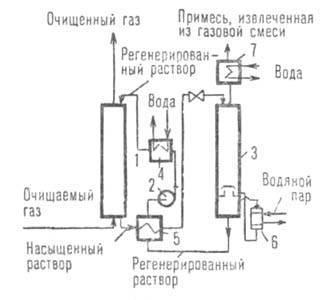
Рис. 4. Принципиальная схема абсорбционно-десорбционного цикла: 1 -абсорбер; 2-насос; 3-десорбер; 4 - холодильник; 5-теплообменник; 6-кипятильник; 7 - конденсатор.
Регенерацию абсорбентов (десорбцию газов) можно проводить снижением давления (вплоть до вакуумирования), нагреванием, отдувкой плохо р-римымигазами и парами кипящего абсорбента, а также сочетанием этих приемов.
Физ. абсорбция осуществляют, как правило, при т-ре окружающей среды (20-40°С) или при пониженных т-рах, т.к. р-римость хорошо р-римых газов возрастает с уменьшением т-ры. Кроме того, при снижении т-ры уменьшается р-римость плохо р-римых газов, т.е. увеличивается селективность и снижаются потери плохо р-римого компонента и загрязнение им извлекаемого газа, а также уменьшаются давление паров абсорбента и его потери. При хим. абсорбции увеличение т-ры приводит к значит. росту коэф. массопередачи и, помимо этого, к возрастанию р-римости мн. абсорбентов в разбавителях, а следовательно, к увеличению до определенного предела общей поглотит. способности абсорбента.
При физ. абсорбции с повышением парциального давления поглощаемого компонента поглотит. способность абсорбента почти всегда увеличивается приблизительно пропорционально парциальному давлению или концентрации газа. Поэтому кол-во циркулирующего абсорбента почти не зависит отконцентрации извлекаемого газа в исходной газовой смеси. При хим. абсорбции характер изменения р-римости газа с ростом его парциального давлениясильно зависит от константы равновесия р-ции и степени превращения абсорбента. В результате при увеличении концентрации извлекаемого газа кол-во циркулирующего абсорбента возрастает.
Физ. абсорбция, как правило, наиб. эффективна при грубой очистке от больших кол-в газа под давлением. Хим. абсорбция чаще всего применяют при извлечении малых кол-в примесей и при тонкой очистке; при этом обычно существенно выше селективность абсорбента, ниже кол-во циркулирующего р-ра вследствие большой поглотит. способности, меньше расход электроэнергии, но выше расход теплоты.
Режим абсорбции. При расчете абсорбции обычно задают параметры очищаемого газа (давление, т-ру, состав) и предъявляют требования к очищенному газу. Необходимый для осуществления абсорбции расход жидкости (кол-во циркулирующего абсорбента) определяется материальным балансом и кинетикой абсорбции. Концентрация у2,в извлекаемого компонента в очищенном газе зависит от х2,в:
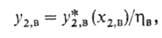
где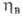 -степень приближения к равновесию на выходе газа из абсорбера, зависящая от скорости процесса. В общем случае:
-степень приближения к равновесию на выходе газа из абсорбера, зависящая от скорости процесса. В общем случае:
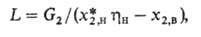
-степень приближения к равновесию на выходе газа из абсорбера, зависящая от скорости процесса. В общем случае:где G2-кол-во извлекаемого газа. При необратимой р-ции миним. кол-во циркулирующего поглотителя определяют из стехиометрич. ур-ния р-ции и находят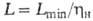 .
.
.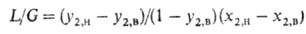
При разомкнутых процессах обычно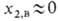 , при циркуляционных - х2,в
, при циркуляционных - х2,в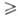 х2,р (концентрация газа в регенерированном р-ре). В простейшем случае (при
х2,р (концентрация газа в регенерированном р-ре). В простейшем случае (при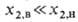 и
и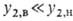 ) имеем:
) имеем: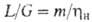 . Если р-римость описывается законом Генри, то
. Если р-римость описывается законом Генри, то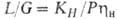 .При этом кол-во циркулирующего абсорбента не зависит от кол-ва извлекаемого газа. При достижении равновесия на выходе из абсорбера
.При этом кол-во циркулирующего абсорбента не зависит от кол-ва извлекаемого газа. При достижении равновесия на выходе из абсорбера
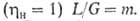
, при циркуляционных - х2,вх2,р (концентрация газа в регенерированном р-ре). В простейшем случае (при и ) имеем:. Если р-римость описывается законом Генри, то.При этом кол-во циркулирующего абсорбента не зависит от кол-ва извлекаемого газа. При достижении равновесия на выходе из абсорбераВажный параметр процесса - т. наз. абсорбционный фактор:
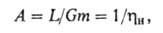
к-рый при полном извлечении газа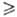 1. Этот фактор равен отношению тангенсов углов наклона рабочей и равновесной линий, к-рые выражают зависимость соотв. реальной и равновесной концентраций извлекаемого компонента в газовой фазе от его концентрации в жидкости. Параметр А одновременно характеризует избыток кол-ва циркулирующего абсорбента по сравнению с минимально необходимым для полного извлечения р-римого газа.
1. Этот фактор равен отношению тангенсов углов наклона рабочей и равновесной линий, к-рые выражают зависимость соотв. реальной и равновесной концентраций извлекаемого компонента в газовой фазе от его концентрации в жидкости. Параметр А одновременно характеризует избыток кол-ва циркулирующего абсорбента по сравнению с минимально необходимым для полного извлечения р-римого газа.
1. Этот фактор равен отношению тангенсов углов наклона рабочей и равновесной линий, к-рые выражают зависимость соотв. реальной и равновесной концентраций извлекаемого компонента в газовой фазе от его концентрации в жидкости. Параметр А одновременно характеризует избыток кол-ва циркулирующего абсорбента по сравнению с минимально необходимым для полного извлечения р-римого газа.Режим десорбции газов (регенерации абсорбентов). Максимально допустимую концентрацию растворенного компонента в регенерированном р-ре определяют из условия равновесия на выходе из абсорбера (при противотоке). Минимально достижимую концентрацию газа
определяют из условия равновесия на выходе из абсорбера (при противотоке). Минимально достижимую концентрацию газа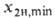 в том же р-ре придесорбции в результате снижения давления, нагревания или отдувки парами абсорбента находят с помощью условия равновесия на выходе р-ра из десорбера:
в том же р-ре придесорбции в результате снижения давления, нагревания или отдувки парами абсорбента находят с помощью условия равновесия на выходе р-ра из десорбера:
определяют из условия равновесия на выходе из абсорбера (при противотоке). Минимально достижимую концентрацию газа в том же р-ре придесорбции в результате снижения давления, нагревания или отдувки парами абсорбента находят с помощью условия равновесия на выходе р-ра из десорбера:где Р и Рпар- общее давление в регенераторе и давление паров абсорбента. В нек-рых случаях, особенно при регенерации хемосорбентов, предельная глубинарегенерации определяется равновесием в к.-л. другом (не в нижнем) сечении десорбера. Это т. наз. критич. сечение определяют после построения равновесной и рабочей линий десорбции либо спец. расчетом.
При регенерации отдувкой плохо р-римым газом предельная глубина регенерации не ограничена давлением и т-рой в десорбере, но зависит, как и при отдувкепарами абсорбента, от расхода отдувочного агента. Его миним. расход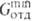 находят из условия соблюдения ур-ния (5) не только на выходе р-ра из десорбера, но и в любом его сечении. Верх, сечение противоточного аппарата, где газ выходит из оегенератора, часто является лимитирующим. Тогда
находят из условия соблюдения ур-ния (5) не только на выходе р-ра из десорбера, но и в любом его сечении. Верх, сечение противоточного аппарата, где газ выходит из оегенератора, часто является лимитирующим. Тогда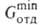 = G2Ф*, где Ф* = р*пар/p2*(t,x2,в)-отношение давления паров абсорбента к давлению газа над р-ром. Если отдувка производится плохо р-римым газом, то Ф * = = (Р — p2,в*)/p2,в. При отдувке парами кипящего р-рителя, когда т-ра в критич. сечении десорбера задана, Ф = = pпар/(Р - pпар). Окончат. расход отдувочного агента можно определить только после построения рабочей и равновесной линий, нахождения местоположения в аппарате критич. сечения и оптимизации абсорбционно-десорбционного цикла.
= G2Ф*, где Ф* = р*пар/p2*(t,x2,в)-отношение давления паров абсорбента к давлению газа над р-ром. Если отдувка производится плохо р-римым газом, то Ф * = = (Р — p2,в*)/p2,в. При отдувке парами кипящего р-рителя, когда т-ра в критич. сечении десорбера задана, Ф = = pпар/(Р - pпар). Окончат. расход отдувочного агента можно определить только после построения рабочей и равновесной линий, нахождения местоположения в аппарате критич. сечения и оптимизации абсорбционно-десорбционного цикла.
находят из условия соблюдения ур-ния (5) не только на выходе р-ра из десорбера, но и в любом его сечении. Верх, сечение противоточного аппарата, где газ выходит из оегенератора, часто является лимитирующим. Тогда = G2Ф*, где Ф* = р*пар/p2*(t,x2,в)-отношение давления паров абсорбента к давлению газа над р-ром. Если отдувка производится плохо р-римым газом, то Ф * = = (Р — p2,в*)/p2,в. При отдувке парами кипящего р-рителя, когда т-ра в критич. сечении десорбера задана, Ф = = pпар/(Р - pпар). Окончат. расход отдувочного агента можно определить только после построения рабочей и равновесной линий, нахождения местоположения в аппарате критич. сечения и оптимизации абсорбционно-десорбционного цикла.При десорбции парами кипящего абсорбента давление, т-ра и концентрация газа в р-ре связаны изобарной равновесной зависимостью Tкип от х2, где Tкип-т-ракипения р-ра при давлении Р в регенераторе. Расчет десорбции смеси газов проводят на основе ур-ния, аналогичного (5):
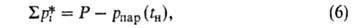
где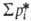 -сумма давлений растворенных газов. Режим десорбции находят совместным решением ур-ния (6) с ур-ниями материального баланса по каждому компоненту методом последоваг. приближений. Равновесную линию десорбции строят по ур-нию:
-сумма давлений растворенных газов. Режим десорбции находят совместным решением ур-ния (6) с ур-ниями материального баланса по каждому компоненту методом последоваг. приближений. Равновесную линию десорбции строят по ур-нию:
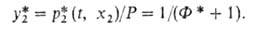
-сумма давлений растворенных газов. Режим десорбции находят совместным решением ур-ния (6) с ур-ниями материального баланса по каждому компоненту методом последоваг. приближений. Равновесную линию десорбции строят по ур-нию:Построение рабочей линии при десорбции парами кипящего абсорбента значительно отличается от построения рабочей линии абсорбера и заключается в совместном решении ур-ний материального и теплового балансов по участкам аппарата при заданном общем расходе теплоты. Рабочая и равновесная линии при десорбции (рис. 5) могут пересечься не в конечных точках х2,в или х2,н, как при абсорбции, а в промежуточном (критическом) сечении десорбера. Это характерно для "сильных" хемосорбентов (напр., при десорбции СО2 из водных р-ров моноэтаноламина) при их глубокой регенерации. Миним. расход отдувочного агента определяется равновесием в критич. сечении и зависит от глубины регенерации.
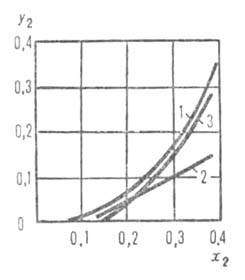
Рис. 5. Равновесная (/) и рабочие (2, 3) линии десорбции СО2 из водного раствора моноэтаноламина при давлении 0,18 МПа и разл. глубине регенерации: x2,у2-концентрации СО2 соотв. в газе и жидкости.
Др. параметры десорбции, в частности число единиц переноса, рассчитывают так же, как при абсорбции. Однако коэф. массопередачи могут неск. отличаться от величин, найденных при абсорбции, вследствие наличия пузырьков газа (пара), возникающих при кипении жидкости или в результате сброса давления, влияния теплоты конденсации отдувочного агента, существенного изменения расходов фаз по высоте аппарата. Скорость хим. абсорбции зависит от скорости прямой р-ции образования соединения между поглощаемым газом и активной частью хемосорбента, а скорость десорбции-от скорости разложения этого соединения и т.п.
Расход энергии на абсорбционное разделение газовых смесей. Этот расход складывается из расхода электроэнергии на циркуляцию р-ра, подачу отдувочного и охлаждающего (воды или воздуха) агентов, рециркуляцию газовых потоков и расхода теплоты. При циклич. процессах физ. абсорбции в основном потребляется электроэнергия на перекачивание р-ра, а при хим. абсорбции -теплота на его регенерацию. При регенерации р-ра теплота расходуется на его нагревание (Qнагр)" покрытие теплоты десорбции Qдес (численно равной теплоте абсорбции) и на создание отдувочного пара (Qотд), если отдувка осуществляется парами кипящего абсорбента:
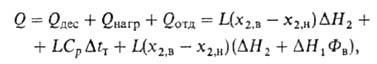
где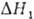 и
и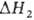 -тепловые эффекты соотв. испарения абсорбента и десорбции;
-тепловые эффекты соотв. испарения абсорбента и десорбции;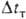 -разность т-р регенерированного и насыщ. р-ров на холодном концетеплообменника. Давление десорбции может сильно сказываться на расходе энергии, особенно при регенерации под вакуумом. При десорбции парамикипящего абсорбента соответствующие изменения давления и т-ры вызывают изменение Ф*. Если
-разность т-р регенерированного и насыщ. р-ров на холодном концетеплообменника. Давление десорбции может сильно сказываться на расходе энергии, особенно при регенерации под вакуумом. При десорбции парамикипящего абсорбента соответствующие изменения давления и т-ры вызывают изменение Ф*. Если
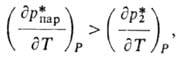
и-тепловые эффекты соотв. испарения абсорбента и десорбции;-разность т-р регенерированного и насыщ. р-ров на холодном концетеплообменника. Давление десорбции может сильно сказываться на расходе энергии, особенно при регенерации под вакуумом. При десорбции парамикипящего абсорбента соответствующие изменения давления и т-ры вызывают изменение Ф*. Еслито с увеличением т-ры Qотд возрастает. В противном случае Qотд уменьшается и при заданном расходе теплоты с увеличением давления десорбциирегенерация не ухудшается, а улучшается, однако до нек-рого предела, определяемого термохим. устойчивостью абсорбента и возрастанием Qнагр. Энергетич. затраты на десорбцию газов (регенерацию абсорбентов), как правило, значительно превышают расход энергии на абсорбцию.
АБС-ПЛАСТИК (луран, люстран, силак, сиколак, терлуран), термопластичный тройной сополимер акрилонитрила, бутадиена и стирола (назв. пластикаобразовано из начальных букв наименований мономеров). Представляет собой дисперсную систему: непрерывная фаза (матрица)-статистич. сополимер стирола с акрилонитрилом мол. м. 120-180 тыс., дисперсная фаза-бутадиеновый, бутадиенстиролъный или бутадиен-нитрильный каучук с размером частиц 0,5-2 мкм. Частицы каучука содержат окклюдированные микрочастицы матричного сополимера и привитые к каучуку макромолекулы этого сополимера, обеспечивающие межфазное взаимодействие. Доля дисперсной фазы-15-30% от массы пластика.
АБС-пластик-непрозрачный, обычно темноокрашенный материал, обладающий высокими влаго-, масло-, кислото-и щелочестойкостью, устойчивостью к действию орг. р-рителей. По мех. прочности, ударной вязкости, теплостойкости и жесткости превосходит ударопрочный полистирол. Атмосферостойкостьпластика относительно невысока, что обусловлено присутствием в макромолекуле каучука ненасыщ. связей. Повышение атмосферостойкости достигается заменой полибутадиена на насыщ. эластомер, напр. бутилакрилатный (ААС-пластик), бутилкаучук, двойной этилен-пропиленовый, хлориров. полиэтилен. Прозрачную модификацию пластика получают, используя 4-й мономерметилметакрилат (при этом повышается и атмосферостойкость сополимера).
Наполнение АБС-пластика короткими стеклянными волокнами (15-30% по массе) приводит к повышению его прочности при растяжении, сжатии и изгибе, росту модуля упругости (в 1,5-2 раза), но к снижению ударной вязкости. Широко применяют вспенивание пластика (при этом плотность снижается обычно на 25-40%) или наполнение его высокодисперсными в-вами.
Нек-рые характеристики АБС-пластика приведены ниже:
Плотн., г/см3 | 1,05-1,08 |
Т. размягч., °С | 90-105 |
Прочность при растяжении, МПа | 35-50 |
Относит. удлинение, % | 10-25 |
Модуль при деформации изгиба, ГПа | 1.5-2,4 |
Ударная вязкость по Шарли (с надрезом), кД.ж/м2 | 10-30 |
Твердость по Бринеллю, МПа | 90-150 |
Т. самовоспл., °С | 395 |
КПВ (нижний) пылевоздушной смеси, г/м3 | 16 |
Методы получения АБС-пластика основаны на радикальной сополимеризации стирола с акрилонитрилом в присут. латекса каучука. При соотношении стирол:акрилонитрил, равном 76:24 (по массе), получают сополимер такого же состава. При др. соотношениях мономеров требуется тщательный контроль однородности образующегося сополимера. Кроме того, с увеличением кол-ва акрилонитрила резко повышается вязкость системы. Наиб. распространение получила двухстадийная эмульсионная сополимеризация по непрерывной или периодич. схеме. На первой стадии синтезируют латекс, на второй - прививают ккаучуку эмульгированные в латексе мономеры. Латекс коагулируют, отделяют от воды и сушат. Образующийся порошкообразный продукт иногда гранулируют.
Пластики с повыш. ударной вязкостью получают обычно в комбиниров. процессе, к-рый сначала ведут в эмульсии или р-ре, а затем - в водной суспензии, что позволяет вводить дополнит. кол-ва каучука. Реализован также трехстадийный синтез пластика в массе; процесс обрывают при содержании в системе 70-80% целевого продукта.
Перерабатывают АБС-пластик литьем под давлением, экструзией. Хладотекучесть пластика позволяет также формовать его при высоких давлениях ниже т-ры стеклования. В произ-ве изделий широко применяют тиснение, печатание и гальванизацию пов-сти.
АБС-пластик-материал для изготовления крупных деталей автомобилей (напр., приборных щитков, элементов ручного управления), корпусов теле- и радиоаппаратуры, телефонов, деталей электроосветит. и электронных приборов, спортинвентаря, мебели, изделий сантехники. Его используют также какнаполнитель, повышающий ударопрочность или (и) улучшающий перерабатываемость композиций на основе ПВХ, поликарбонатов, полистирола. Мировое произ-во АБС-пластика-ок. 2 млн. т/год (1980).
(+)-АБСЦИЗОВАЯ КИСЛОТА [АБК; абсцизин II; дормин; (S)-(Z, Е)-3-метил-5-(1 -гидрокси-4-оксо-2,6,6-триметил-2-циклогексенил)-2,4-пенталиеновая к-та], бесцв. кристаллы; т. пл. 160-162°С,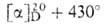 (метанол); раств. в ацетоне, эфире, хлороформе, этилацетате, в воде малорастворима. В УФ-спектре
(метанол); раств. в ацетоне, эфире, хлороформе, этилацетате, в воде малорастворима. В УФ-спектре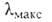 260 нм,
260 нм,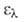 21 500. АБК выделяют обычно из плодов и листьев растений, где она содержится в кол-вах 10-9 - 10-6 мг/г сырой массы. Фотохим.окислением 3-метил-5-(2,6,6-триметил-1,3-циклогексадиенил)-цис, транс-2,4-пентадиеновой к-ты получают рацемат с т. пл. 191°С.
21 500. АБК выделяют обычно из плодов и листьев растений, где она содержится в кол-вах 10-9 - 10-6 мг/г сырой массы. Фотохим.окислением 3-метил-5-(2,6,6-триметил-1,3-циклогексадиенил)-цис, транс-2,4-пентадиеновой к-ты получают рацемат с т. пл. 191°С.
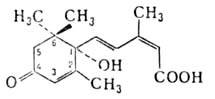
(метанол); раств. в ацетоне, эфире, хлороформе, этилацетате, в воде малорастворима. В УФ-спектре260 нм,21 500. АБК выделяют обычно из плодов и листьев растений, где она содержится в кол-вах 10-9 - 10-6 мг/г сырой массы. Фотохим.окислением 3-метил-5-(2,6,6-триметил-1,3-циклогексадиенил)-цис, транс-2,4-пентадиеновой к-ты получают рацемат с т. пл. 191°С.АБК-наиб. распространенный и важный прир. ингибитор роста растений (антагонист ауксинов, гиббереллинов и цитокининов), ускоряет созревание и старение растений, опадение листьев и плодов, обеспечивает переход растений в состояние покоя. Биосинтез АБК происходит путем конденсации трех молекул (+ )-мевалоновой (3-метил-3,5-дигидроксивалериановой) к-ты с отщеплением СО2 и Н2О под действием АТФ и ряда ферментов. АБК, вероятно, образуется также окислит. расщеплением каротиноидов. Она находится в растениях как в своб. виде, так и в форме малоактивного конъюгата с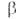 -D-глюкозой-(+)-абсцизил-
-D-глюкозой-(+)-абсцизил-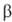 -D-глюкопиранозида, из к-рого может высвобождаться при гидролизе. Дезактивация АБК происходит в осн. в результате окисления одной из метальных групп в положении 6 до СН2ОН с послед. замыканием СН2О-мостика в положениях 6 и 2 кольца, что приводит к образованию фазеевой к-ты.
-D-глюкопиранозида, из к-рого может высвобождаться при гидролизе. Дезактивация АБК происходит в осн. в результате окисления одной из метальных групп в положении 6 до СН2ОН с послед. замыканием СН2О-мостика в положениях 6 и 2 кольца, что приводит к образованию фазеевой к-ты.
-D-глюкозой-(+)-абсцизил--D-глюкопиранозида, из к-рого может высвобождаться при гидролизе. Дезактивация АБК происходит в осн. в результате окисления одной из метальных групп в положении 6 до СН2ОН с послед. замыканием СН2О-мостика в положениях 6 и 2 кольца, что приводит к образованию фазеевой к-ты.
Комментариев нет:
Отправить комментарий